|
|
You are here: Foswiki>Química Web>Docsoftwareqt>DocqtAmber (23 May 2023, AdminUser)Edit Attach
-- AdminUser - 30 Jan 2023
Que es AMBER?
AMBER es el acrónimo de "Assisted Model Building with Energy Refinement" con el que se nombra al conjunto de herramientas compuestas por Amber18 , principalmente el programa de simulacion pmemd (“pmemd” stands for “Particle Mesh Ewald Molecular Dynamics”). y AmberTools18, un conjunto de programas con licencia no propietaria para análisis y simulación biomolecular. AMBER18 integra el uso de GPU`s para los métodos GPU accelerated Thermodynamic Integration (TI) y Free Energy Perturbation (FEP).
 Para ejecuitar el container de Amber usted debe informar a singularity el directorio de trabajo y/o donde estan ubicados sus datos(ruta o path) usando la variable "SINGULARITY_BINDPATH", P. eje. los datos de este tutorial están en "/media/home/jrbolivarm", por lo que debo pasar esta ruta a la variable:
Para ejecuitar el container de Amber usted debe informar a singularity el directorio de trabajo y/o donde estan ubicados sus datos(ruta o path) usando la variable "SINGULARITY_BINDPATH", P. eje. los datos de este tutorial están en "/media/home/jrbolivarm", por lo que debo pasar esta ruta a la variable:
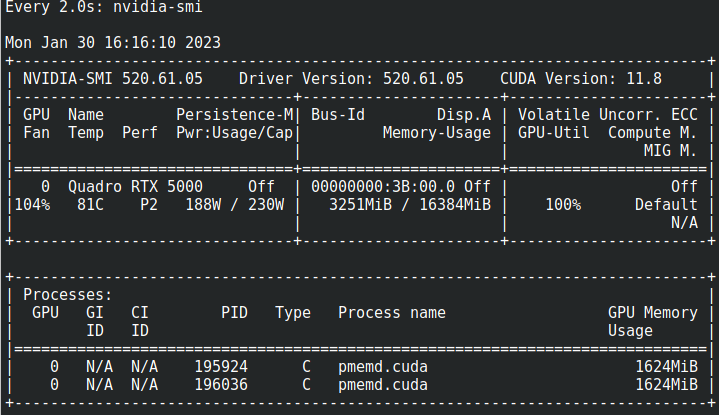
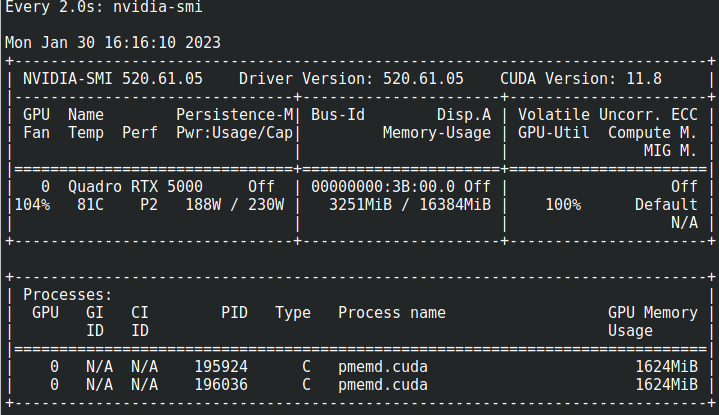
 El uso de la CPU puede visualizarse con "top", el uso de la GPU puede verse con "nvidia-smi"
El uso de la CPU puede visualizarse con "top", el uso de la GPU puede verse con "nvidia-smi"
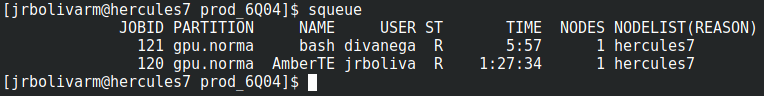
 En la federación de Clusters del CECC los recursos compartidor entre usuarios son aportados por cluster asociados, para garantizar un uso justo, todos deben realizar el envio de trabajos a través del sistema por lotes que ejecutará las aplicaciones con los recursos reservados.
En la federación de Clusters del CECC los recursos compartidor entre usuarios son aportados por cluster asociados, para garantizar un uso justo, todos deben realizar el envio de trabajos a través del sistema por lotes que ejecutará las aplicaciones con los recursos reservados.
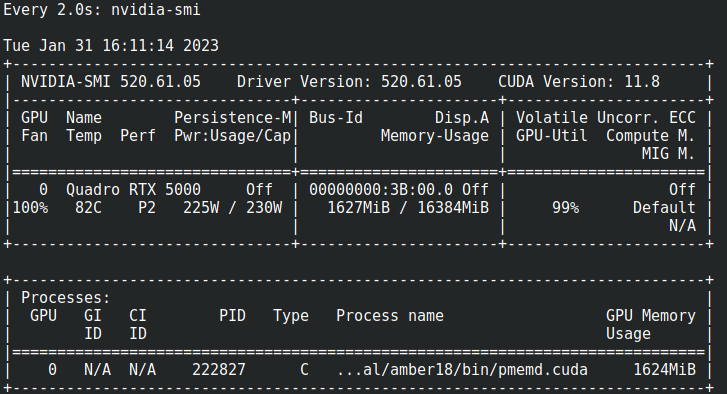
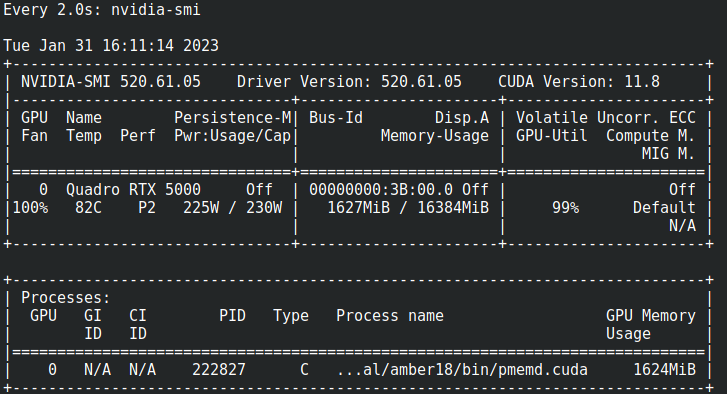
 Tambien puede usar nvidia-smi para ver el uso de la GPU
Tambien puede usar nvidia-smi para ver el uso de la GPU
AMBER es el acrónimo de "Assisted Model Building with Energy Refinement" con el que se nombra al conjunto de herramientas compuestas por Amber18 , principalmente el programa de simulacion pmemd (“pmemd” stands for “Particle Mesh Ewald Molecular Dynamics”). y AmberTools18, un conjunto de programas con licencia no propietaria para análisis y simulación biomolecular. AMBER18 integra el uso de GPU`s para los métodos GPU accelerated Thermodynamic Integration (TI) y Free Energy Perturbation (FEP).
El container con Amber18 esta en: /localapps/Amber18_cuda_10.2_centos7Usted puede ejecutar Amber18 en la Federación de cluster en una sesión iteractiva donde los resultados se esperan en tiempos NO MAYORES a Cuatro(4) horas; para tiempos superiores debe agendar la ejecucion mediante scripts.
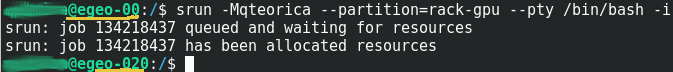
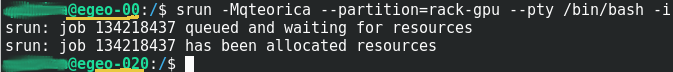
Ejecutar Amber18 en una sesion Interactiva vía SRUN en un cluster Federado
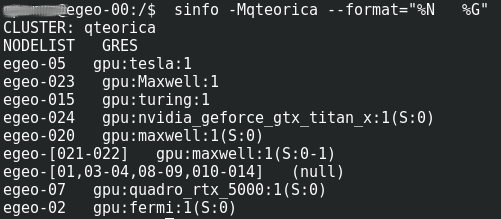
Este procedimiento posibilita usar inmediatamente los recursos con los que ejecutará los procesos; Siempre y cuando esten disponibles y su programa no requiera mas de Cuatro(4) horas de reloj pared – wall-clock – de procesamiento. Luego de conectarse con una sesión ssh en el nodo de logeo del cluster o en el nodo de logeo de la Federación de clusters, debe solicitar los recursos en una sesión interactiva usando el commando srun con los parametros| Commando | Parametro Formato Corto y Largo | Ejemplo | Descripcion |
| srun | -M NombreCluster -clusters= NombreCluster |
-Mqteorica --clusters=qteorica | Cluster Federado donde estan los Recursos |
| -pNombre_de_Particion --partition=Nombre_de_Particion | -prack-gpu --partition=rack-gpu | Particion del cluster federado donde estan los recursos |
srun -Mqteorica --partition=rack-gpu --pty /bin/bash -i
Para ejecuitar el container de Amber usted debe informar a singularity el directorio de trabajo y/o donde estan ubicados sus datos(ruta o path) usando la variable "SINGULARITY_BINDPATH", P. eje. los datos de este tutorial están en "/media/home/jrbolivarm", por lo que debo pasar esta ruta a la variable:
export SINGULARITY_BINDPATH="/media:/media"Todo el árbol y contenido del directorio "/media" del sistema operativo, sera accesible en la ruta "/media" dentro del container. Luego puede ejecutar una "shel"l dentro del container y acceder a los datos en "/media/home/jrbolivarm" usando la GPU(--nv)
El uso de la CPU puede visualizarse con "top", el uso de la GPU puede verse con "nvidia-smi"
En la federación de Clusters del CECC los recursos compartidor entre usuarios son aportados por cluster asociados, para garantizar un uso justo, todos deben realizar el envio de trabajos a través del sistema por lotes que ejecutará las aplicaciones con los recursos reservados.
Usar un script para correr Amber18 en un HPC del CECC
Para enviar su trabajo a procesar en un HPC del CECC debe construir un script de shell con algunas directivas que especifican la cantidad de CPU, memoria, tiempo a usar, número de nodos y otras directivas que el sistema SLURM interpretará al enviarlo con el comando sbatch . Para ejecutar el mismo ejemplo iteractivo de Amber18 cree el archivo run_amber.sh, incluya la ruta de los directorios a usar con la variable "SINGULARITY_BINDPATH", en el ejemplo, los datos estan en "/media/home/jrbolivarm". Recuerde que el directorio de trabajo(donde se ubica los archivos/directorios de entrada/salida) por omision es el directorio actual de trabajo(pwd) y tambien debe ser incluido la variable "SINGULARITY_BINDPATH".#!/bin/bash -l
#SBATCH --job-name=AmberTE # Nombre del proceso...se mostrara en la cola de procesos
#SBATCH --gres=gpu:turing:1 # GPU por Nodo
#SBATCH --partition=gpu.normal.q # Particion o Grupo donde se encuentra el nodo a usar
#SBATCH --cpus-per-task=6 # CPU cores/threads
#SBATCH --output=amber18_slurm_%j.out # donde almacenara la salida estandar (%j es el ID del proceso o JOBID)
#SBATCH --error=amber18_slurm_%j.err # donde almacenara la salida del error estandar(%j es el ID del proceso o JOBID)
export SINGULARITY_BINDPATH="/media:/media"
singularity run --nv /localapps/Amber18_cuda_10.2_centos7 pmemd.cuda -O -i prod.in -o prod.out -p 6Q04_ion.prmtop -c equil.rst7 -r prod.rst7 -inf prod.info -x prod_6Q04_ion.nc
Luego, para agendar los recursos solicitados y correr el proceso ejecuite en la consola o terminal:
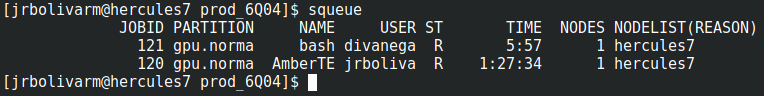
* _sbatch_* run_amber.shPuede ver que su proceso corre en la cola de tareas SLURM con el comando squeue
Tambien puede usar nvidia-smi para ver el uso de la GPU


| I | Attachment | Action | Size | Date | Who | Comment |
|---|---|---|---|---|---|---|
| |
egeoGRES1.png | manage | 54 K | 22 May 2023 - 23:53 | AdminUser | egeoGRES1 |
| |
egeoGRES2.png | manage | 28 K | 23 May 2023 - 00:52 | AdminUser | egeoGRES2 |
| |
srun1.png | manage | 10 K | 02 Feb 2023 - 08:38 | AdminUser | srun1.png |
| |
srun2.png | manage | 27 K | 02 Feb 2023 - 08:39 | AdminUser | srun2.png |
| |
srun3.png | manage | 50 K | 02 Feb 2023 - 08:40 | AdminUser | srun3.png |
| |
srun4.png | manage | 24 K | 02 Feb 2023 - 08:40 | AdminUser | srun4.png |
| |
srun5.png | manage | 47 K | 02 Feb 2023 - 08:41 | AdminUser | srun5.png |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Edit | Attach | Print version | History: r7 < r6 < r5 < r4 | Backlinks | View wiki text | Edit wiki text | More topic actions
Topic revision: r7 - 23 May 2023, AdminUser
- Toolbox
-
 Create New Topic
Create New Topic
-
 Index
Index
-
 Search
Search
-
 Changes
Changes
-
 Notifications
Notifications
-
 RSS Feed
RSS Feed
-
 Statistics
Statistics
-
 Preferences
Preferences
Ideas, requests, problems regarding Foswiki? Send feedback